Human genome assemblies with nanopore, an update
We recently participated in a collaborative effort to sequence, assemble, and analyze a human genome (GM12878) using the Oxford Nanopore MinION (Jain et al. 2018). Since then, we’ve also developed a trio-based strategy for assembling complete haplotypes from long-read data (Koren et al. 2018). Oxford Nanopore has continued to advance in the meantime, releasing several major base-calling updates. Other tools, such as Nanopolish, have also gotten faster and added new functionality, like methylation-aware polishing. So, we decided to re-analyze the dataset from the paper using the latest base calling and assembly tools. The new assembly increases the NG50 to over 10 Mbp and trio binning accurately reconstructs key MHC genes for both haplotypes.
We started by re-calling the raw data using Albacore v2.1 with default parameters. The total coverage increased from 37X to 41X. A free 4-fold coverage increase from a software update, not bad! The average read length also increased from 7.3 to 8.1 kbp. Re-assembling with Canu 1.6 gave an improved NG50 of 10.2 Mbp and required approximately 150k cpu hours, as in the paper. We also assembled the genome using a combination of Canu 1.7 read correction with WTDBG contigging. This further increased the NG50 to 12.4 Mbp due to improved read correction in Canu 1.7, and required 30k cpu thanks to the speed of WTDBG. There is still room for improvement with coverage or longer reads since the most continuous human assemblies are over 20 Mbp. The Cliveome is currently the most continuous, with an NG50 of 29 Mbp from a high-coverage combination MinION and GridION data.
The Canu + WTDBG assembly is more continuous than either a Miniasm assembly (NG50=6.7Mbp, 8k cpu with Racon) or a WTDBG-only assembly (NG50=8.7Mbp, 0.5k cpu), neither of which perform read correction. Thus, Canu’s read correction does appear to benefit assembly and we plan to make this Canu + WTDBG pipeline available in a future Canu release. In the meantime, users can run the Canu correction module and feed its output directly into WTDBG for a faster assembly option.
We also evaluated the base quality of the new Canu + WTDBG assembly. The identity is 98.94%, up from the 95.94% we previously reported. This also improves upon the 97.80% identity we reported for a chromosome 20 assembly of Scrappie-called reads (Jain et al. 2018). After two rounds of Nanopolish with its new “CpG methylation” mode, the Canu + WTDBG assembly identity improved to 99.76%. We still see a deletion bias and a high fraction of short indels, but a peak for Alu indels is clearly visible now, whereas it was not before. We are encouraged by these improvements.
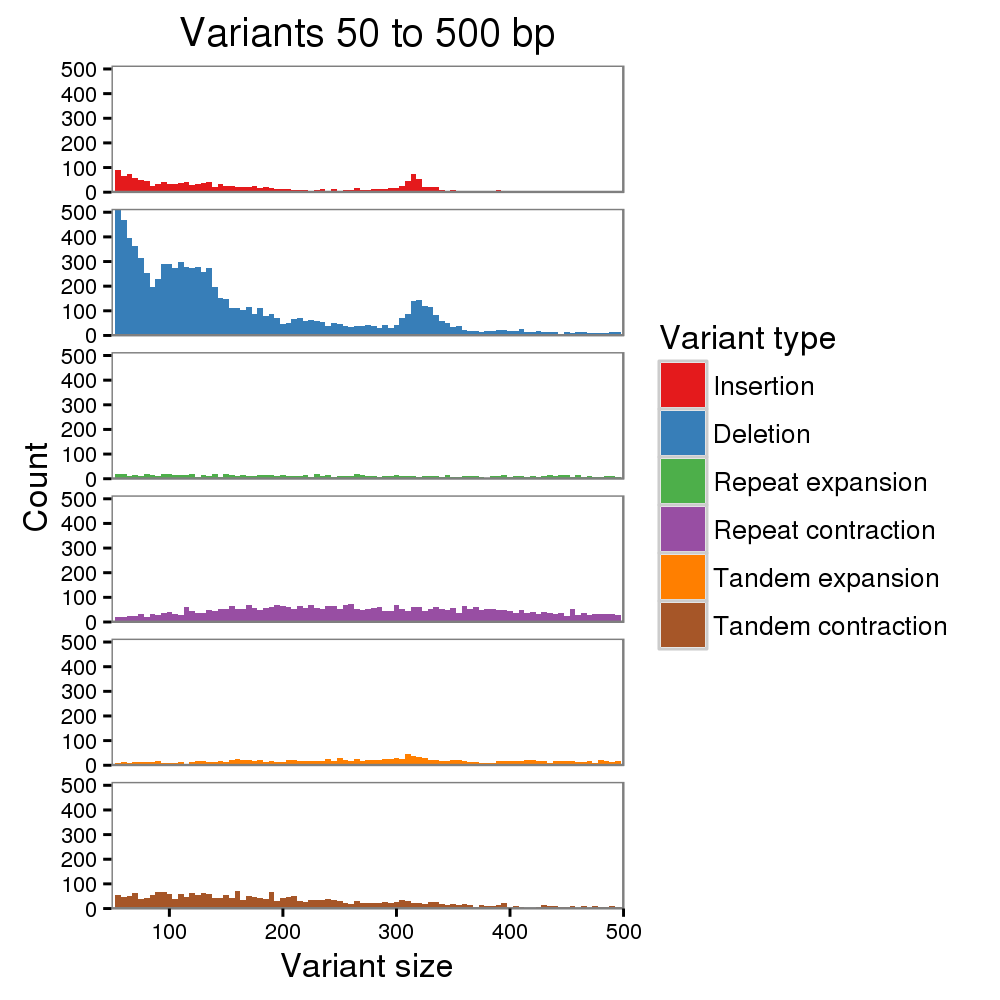
However, 99.76% is still a bit disappointing. It is much better than 95.94%, but represents an error about every 400 bp. Commonly, Illumina data is used to polish away this remaining 0.25% of error, but this has its own issues, especially for heterozygous genomes and complex repetitive regions where short-read mapping is a challenge.
Because parental data is available for GM12878, we also attempted complete haplotype assembly using our recently described trio binning approach (Koren et al. 2018). Using TrioCanu, we classified the nanopore reads for GM12878 into maternal and paternal haplotype bins prior to assembly. Despite the lower coverage (40x Nanopore vs. 70x PacBio), we saw a similar classification rate (85% of bases classified by haplotype) and similar NG50 stats for the assembled haplotigs (1.3 Mbp paternal, 1.2 Mbp maternal). The identity for both haplotypes after two rounds of CpG Nanopolish was 99.24%. Consistent with our results on other datasets, we would expect both larger NG50s and higher identity with more coverage. In this case, Nanopolish is dealing with less than 20x coverage per haplotype.
We also aligned the two nanopore haplotypes to one another to call structural variants and compared these results to the same analysis performed with PacBio:
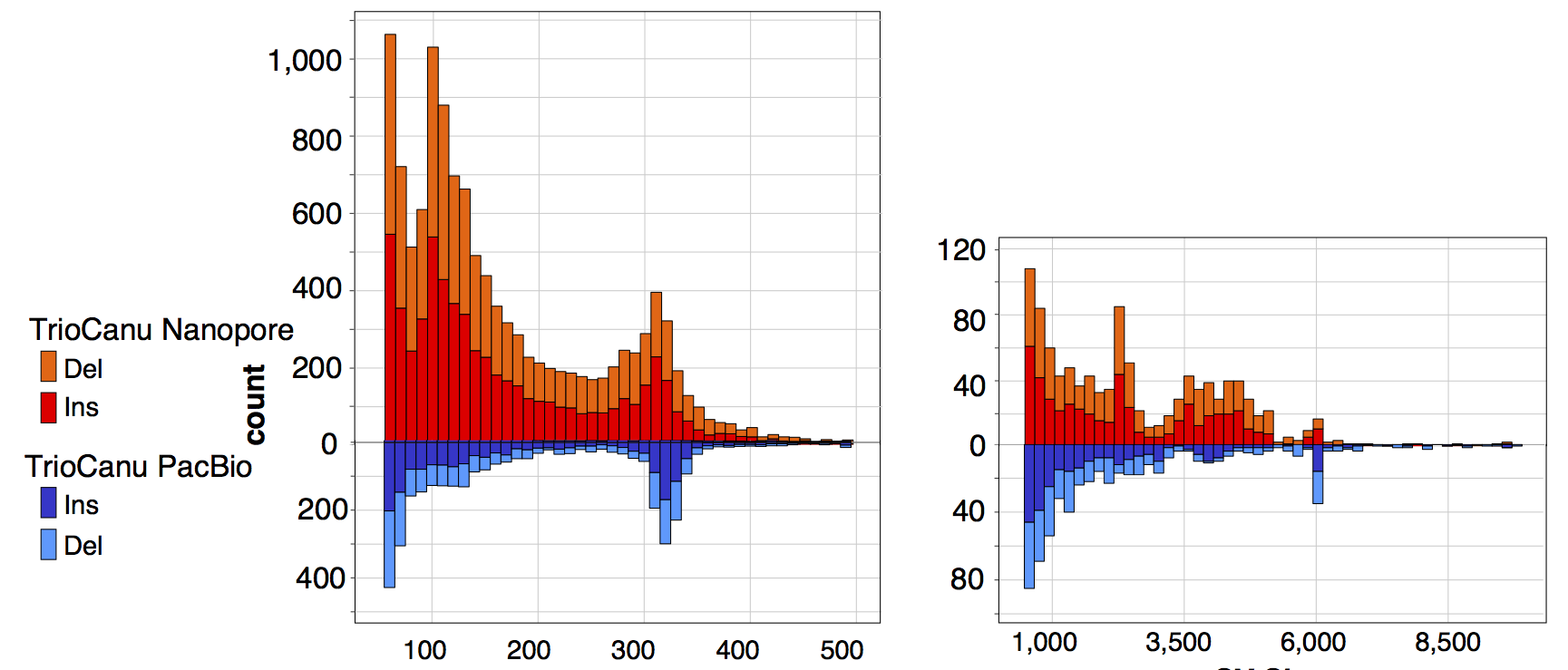
There are again more short indels than expected in the Nanopore assembly (presumably due to base-calling biases and the lower depth of coverage). In comparison, the PacBio haplotypes show more concentrated SV peaks at the typical Alu and LINE sizes (300bp and 6kb). Repeating the MHC analysis from our trio binning preprint, the HLA typing genes in both nanopore haplotypes are correct at G-level resolution and properly phased, with an average edit distance of 1 bp per gene (12 errors total). This is better than the 10x Genomics result but worse than the PacBio result presented in the preprint.
Highlighting the difficulty of polishing complex regions with Illumina data, attempting to run Pilon on each nanopore haplotype using the parental Illumina data actually reduced the quality and introduced additional errors in several MHC genes. However, restricting Pilon to only correct indels did fix all typing gene errors and yielded a final consensus accuracy of 99.92%. (Note that this experiment was only for evaluative purposes, and naive polishing with parental data runs the risk of masking de novo variants in the child.)
Conclusion
Oxford Nanopore continues to make impressive strides. Recent software improvements (primarily for base calling) have almost doubled the assembly NG50 size for the GM12878 assembly, without the addition of any new data. In parallel, we are continuing to work on Canu performance and in the meantime recommend Canu + WTDBG as good compromise between speed and accuracy. Extremely continuous assemblies are clearly possible using long-read nanopore data alone. The last remaining hurdle is final consensus accuracy, which continues to come up short of PacBio and requires additional polishing using complementary data (e.g. Illumina) to reach “reference” quality.
Nanopore data also appears well-suited for trio binning. For long-read reference genome assembly, we now suggest collecting parental Illumina data wherever possible. This approach greatly simplifies the assembly of heterozygous genomes and yields accurate representations of both haplotypes. If performed on a hybrid cross, this method will yield two reference genomes from a single sequencing project, as we recently demonstrated using a cattle F1 cross. Human trios can be harder to come by, but the method is applicable and works well for reconstructing heterozygous structural variation.
Finally, a note of caution on Illumina polishing with Pilon. While it can improve consensus statistics overall, it can worsen the assembly in some regions, especially complex repetitive sequence like the MHC. If using Pilon, we recommend limiting the allowable edits and focusing on the primary nanopore error mode (indels). Using a purpose-built variant caller instead, such as FreeBayes, is also advisable but requires custom editing of the consensus. A more sophisticated approach to hybrid Nanopore + Illumina assembly would be ideal, but we have not yet seen a satisfactory solution. Ultimately, resolving the last bit of nanopore error without an additional technology would be best, and it will be exciting to see if Oxford Nanopore can achieve this with future updates to sequencing and/or base calling methods.
You can find a description of our assembly methods in the Jain et al. 2017 paper and the trio binning paper Koren et al. 2018. Sergey will be presenting this work at London Calling this week.
Update 2018-05-24: The basecalled data is available from the NA12878 consortium. Update 2018-05-29: We’ve made the basecalled data split into maternal, paternal, and unclassified bins available. Update 2018-10-28: We’ve made the Canu + Nanopolish + Pilon + Racon assembly, which is 99.99% identity, available. Download the Canu 1.7 + WTDBG + Nanopolish assembly or the maternal and paternal assemblies.