MashMap: approximate long-read mapping using minimizers and MinHash
Chirag Jain recently presented a paper at RECOMB’17 titled “A fast approximate algorithm for mapping long reads to large reference databases” (preprint | proceedings). This paper describes the algorithms behind MashMap, which is our new tool designed for approximate read mapping. Chirag joined the lab last year as a summer fellow, and I asked him to write a new read mapper. (How else does one learn bioinformatics?) He clearly lived up to the challenge, and I think the paper contains some useful ideas for the looming “long-read” era. I wanted to summarize those ideas here for anyone who missed RECOMB.
MashMap
When I talk about “long reads”, I am referring to single-molecule sequencing reads from Pacific Biosciences or Oxford Nanopore that can range anywhere from 1 kb to >100 kb in length at ~85% accuracy, or better. This new datatype is incompatible with the existing mapping algorithms designed for short, accurate reads. As we recently discovered when sequencing a human genome with nanopore (Jain et al. 2017), mapping >100 kb nanopore reads to the human genome with BWA can take a while. So, we have been rethinking how to design a mapper for long, noisy reads. MashMap is a work in progress, but successfully implements some of our early ideas. Here are the highlights:
1. A hierarchical minimizer index
The minimizer comes from Mike Roberts and co. who developed the idea for genome assembly (Roberts et al. 2004). Coincidentally, the same idea arose outside of bioinformatics as winnowing a year prior (Schleimer et al. 2003). Simply put, for a fixed k-mer size and an ordering on those k-mers (e.g. lexicographically or by hash value), the minimizer of a string is its smallest k-mer. Typically, a window of size w is slid across a larger string to collect a minimizer for each length w substring. Why is this useful? The minimizer serves as a summary of the window, but in a smaller package. We have demonstrated a tremendous speedup of long-read overlapping using related ideas (Berlin et al. 2015), and Heng Li recently developed a minimizer-based, long-read mapper/overlapper called minimap (Li 2016).
MashMap takes a similar approach, with a few new twists. The basics are to load all minimizers from a reference genome into a hash table and compare a read’s minimizers against this index to find significant hits. Uniquely, MashMap builds a hierarchical minimizer index using multiple window sizes {w, 2w, 4w…}, so that it can adjust to different read lengths on the fly. This allows MashMap to use a larger window size for larger reads, without affecting the accuracy or ‘big-O’ complexity of the algorithm. Because of this, mapping time is independent of read length, and a 100 kb read can be mapped as quickly as a 1 kb read — this is certainly not the case for seed-and-extend algorithms like BLAST. However, I should note that MashMap currently reports only end-to-end mappings for each read, unlike BLAST and others that also report partial (local) alignments.
2. Sequence identity estimate directly from minimizers
The key innovation of MashMap is to estimate the percent identity of a read-to-reference alignment from the minimizers alone. This is a useful feature because the quality of a mapping can be judged without a costly gapped alignment. To accomplish this, we built upon the Mash distance (Ondov et al. 2016), which estimates the mutation rate between two sequences using MinHash (Broder 1997). For MinHash, instead of remembering only the smallest k-mer in a window (the minimizer), you remember the s smallest k-mers (the sketch). As shown in the Mash paper, the identity of two sequences can be estimated directly from their sketches. MashMap extends this idea to the mapping problem, and uses a sketch of minimizers to estimate both read position and identity. The correlation between true sequence identity and MashMap estimates is shown here for PacBio reads mapped to the human genome:
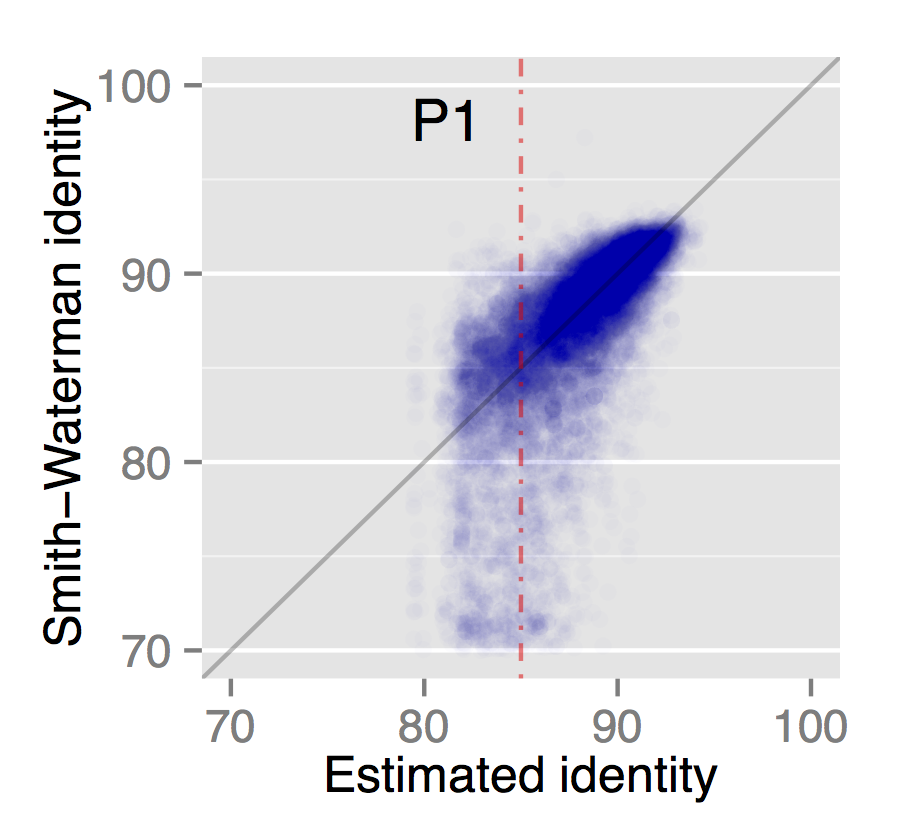
The light smear at the bottom is due to a few repetitive sequences for which MashMap overestimates the identity.
3. Simplified mapping parameters
Ever since writing Nucmer (Delcher et al. 2002), users ask me “What parameters should I use to align genome A to genome B?” and I never have a satisfying answer. This is my biggest frustration with most alignment tools. MashMap attempts to address this by focusing on only two parameters: minimum match length and minimum identity. The goal is to identify all mappings meeting both criteria. Because we now have a nice link between minimizers and identity, the internal parameters of the algorithm can be automatically adjusted to meet the requirements. I find this much more intuitive than the seed length and clustering parameters exposed by tools like Nucmer. As expected, increasing the minimum length and identity requirements improves the performance of the algorithm.
4. Mapping to all RefSeq
In the original Mash paper we clustered the entire NCBI RefSeq database by sequence similarity, which one reviewer called a “heroic” computation. That kind comment inspired us to attempt some more heroics for the MashMap paper, so we set out to map a long-read metagenomic dataset to all of RefSeq. Even without a clever indexing scheme (just a big old hash table), we were able to map ~2 PacBio reads per CPU second to all of RefSeq with good sensitivity. Ultimately, we would like to use such mappings to determine the genomic origin of a sequencing read as it pops out of a nanopore sequencer in real time.
What’s next
Chirag has rejoined the lab for another summer (hooray!) and we plan to expand on this work. I would like to generalize these ideas to the local alignment problem, which would be useful for whole-genome alignment, structural variant detection, and more. Essentially, I would like a fast, approximate, Nucmer replacement. Additionally, I think these ideas could be naturally extended to genome graphs, e.g. do MinHash identity estimation over a de Bruijn graph path to rapidly evaluate mapping quality. Hopefully we will have some more interesting results to share after another summer working together.